
Macauley Lab
Research Focus

The focus of our research is to understand diseases of the central nervous system (CNS) and how risk factors such as metabolic dysfunction, sleep impairment, and vascular damage disrupt healthy brain function to cause neurodegenerative disease. Ultimately, the goal is to leverage these mechanistic findings as therapeutic targets for treating CNS disease.
My work focuses on two main areas: 1) the mechanistic interplay between Alzheimer’s disease (AD) and type-2-diabetes (T2D) and 2) the treatment of neurodegenerative disorders, including AD and lysosomal storage diseases (LSDs). To study AD, my laboratory uses rodent models, non-human primates, and human data to understand how metabolic or vascular perturbations affect the progression of Alzheimer’s-related pathology. For our rodent studies, we use a variety of in vivo techniques, including glucose clamps, in vivo microdialysis, in vivo biosensors, EEG/EMG recordings, and small animal neuroimaging to study the acute effects of metabolic challenges on cerebral metabolism, neuronal activity, Aβ/tau dynamics, and sleep. For our chronic studies, we use rodent models and non-human primates to investigate how Alzheimer’s disease risk factors, like metabolic dysfunction or sleep disruptions, impact Aβ/tau pathology, learning and memory, cerebral metabolism, and brain network connectivity. Our lab is using both pharmacological and non-pharmacological interventions to target metabolic dysfunction to reverse Alzheimer's pathology and rescue sleep.
In addition to my interests in T2D and AD, we study mechanisms of neurometabolism, neurodegeneration, and neuroinflammation in lysosomal storage diseases (LSD) and how targeting different aspects of pathology with combination therapies enhances efficacy. Our findings not only characterized the temporal-spatial spread of CNS disease and functional deficits in mouse models of Niemann-Pick Type A, infantile neuronal ceroid lipofuscinosis, Pompe disease, Krabbe disease, and late-infantile neuronal ceroid lipofuscinosis, but also identified secondary disease mechanisms associated with neurodegeneration that need to be addressed in treatment strategies for these disorders.
Lastly, a significant portion of our laboratory is dedicated to developing novel biomarkers, tools, and technology to improve our studies in the field of Alzheimer's disease. Through a network of collaborations, we are developing novel exosome- and imaging-based biomarkers for staging Alzheimer's disease as well as generating improved mouse and non-human primate models of Alzheimer's disease pathology and resilience.
Alzheimer's disease and type-2-diabetes

Metabolic dysfunction is central to the development of Alzheimer’s disease. Moreover, epidemiological studies suggest that patients with type 2 diabetes (T2D) have an increased risk for developing Alzheimer’s disease (AD). Therefore, a large focus of our laboratory is how metabolic dysfunction, like glycemic variability and glucose intolerance, affects the production, clearance, and aggregation of Aβ and tau (Macauley et al, 2015; Stanley, Macauley et al, 2016; Harris et al, 2016; Kavanagh et al, 2019).
Our initial work investigated whether hyperglycemia, or elevated blood glucose levels, or hyperinsulinemia, elevated blood insulin levels, increased amyloid-β (Aβ) levels in the brain's interstitial fluid (ISF). By coupling in vivo microdialysis and glucose clamps, we developed a novel approach to dynamically modulate systemic blood glucose and/or insulin levels while sampling proteins and metabolites within the brain’s interstitial fluid (ISF) in unanesthetized, freely moving mice. We found that hyperglycemia increased Aβ production in the hippocampus through an activity dependent mechanism; an effect that is exacerbated in mice with amyloid plaques. Interestingly, hyperinsulinemia did not have the same effect, suggesting hyperglycemia is a more potent driver of Aβ production than hyperinsulinemia. We also found a direct correlation between ISF glucose and ISF Aβ concentrations, providing a causal relationship between T2D and AD. Recently, we extended these studies to non-human primates to demonstrate the same phenomenon- peripheral hyperglycemia, not hyperinsulinemia, in vervet monkeys with type-2-diabetes correlates with decreased CSF Aβ40 and Aβ42, a biomarker of Alzheimer’s disease pathology in the brain.
We continue to explore how changes in peripheral or cerebral metabolism affect brain excitability and Aβ/tau metabolism. First, our initial work demonstrated that hyperglycemia modulates extracellular concentrations of amyloid-β (Aβ) in an activity-dependent manner by altering the activity of inward rectifying, ATP-sensitive potassium (KATP) channels. Using pharmacological and genetic approaches, we are exploring the role of KATP channel activity in Alzheimer's disease. Second, our work investigates how hyperglycemia impacts neuronal activity, synaptic plasticity and network connectivity during healthy aging. Third, we are exploring how hyper- and hypoglycemia affects cerebral metabolism, brain excitability, and Aβ/tau pathology in order to further understand the complex relationship between T2D and AD. Lastly, we are exploring why regions vulnerable to Aβ deposition are uniquely reliant on glucose compared to regions resilient to Aβ pathology. Through collaborative studies, we found that alterations in key glycolytic enzymes involved in aerobic glycolysis are altered as a function of age and Aβ, impacting learning and memory differentially. The ultimate goal of our work is to elucidate the role of glucose metabolism in Alzheimer's disease.
Sleep, metabolism, and Alzheimer's disease



Carroll & Macauley, 2019
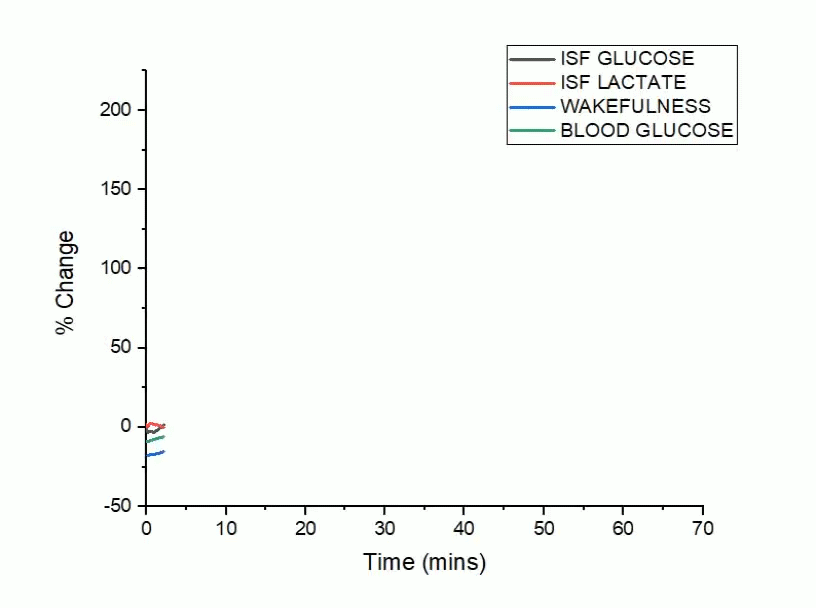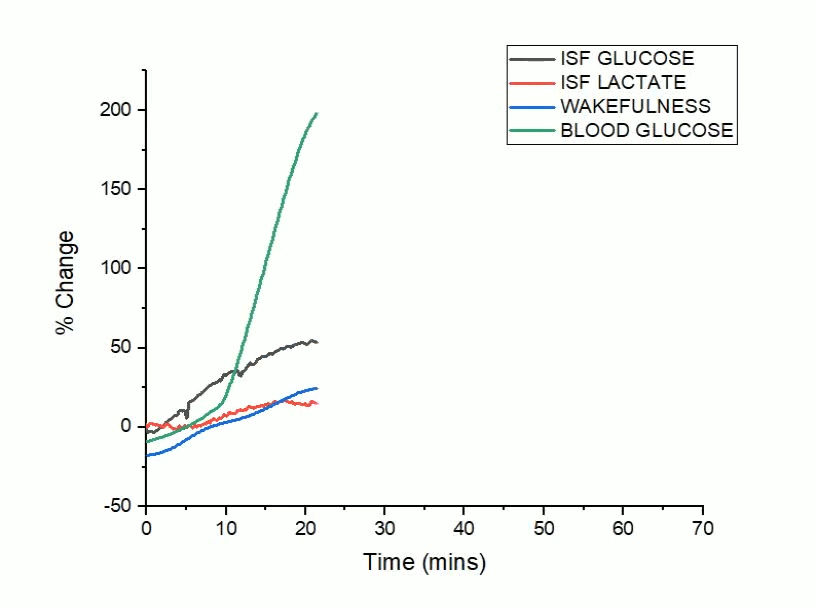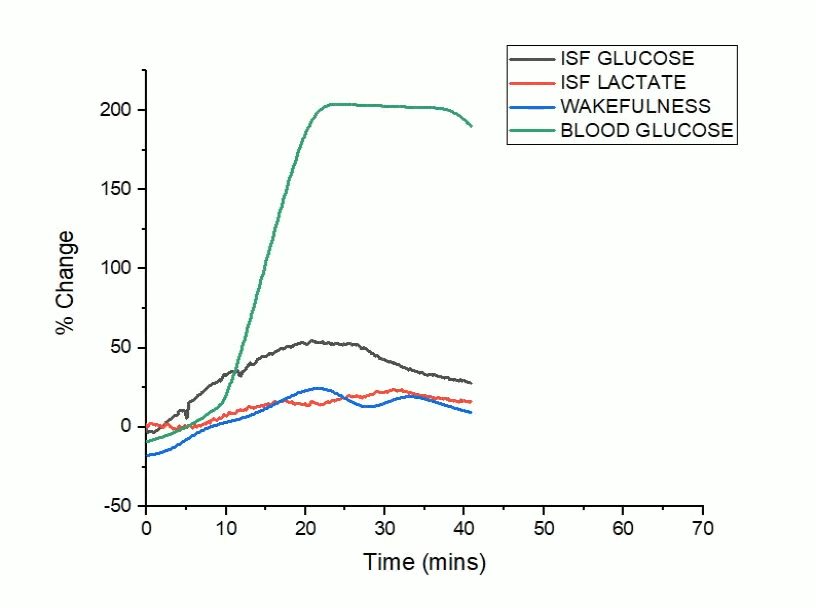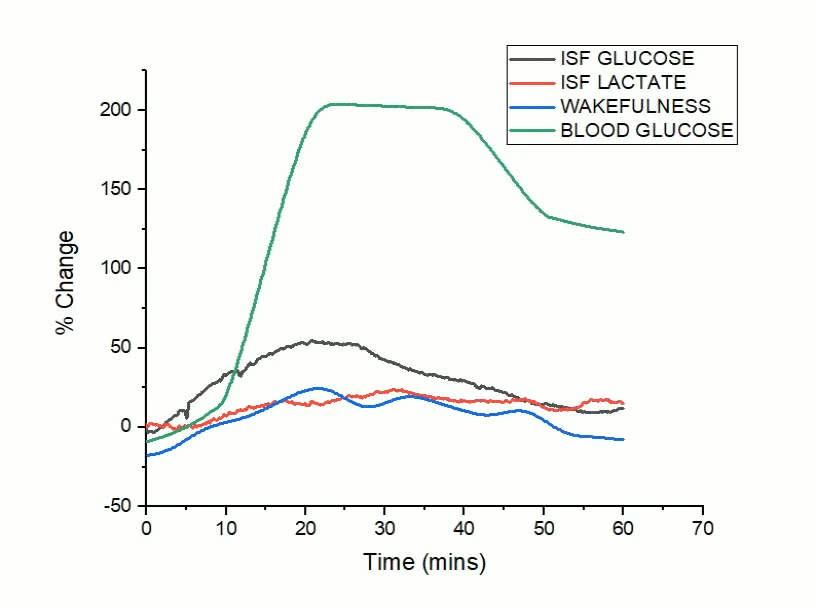
A bidirectional relationship exists between Alzheimer’s disease and sleep, where disrupted sleep increases amyloid-beta (Aβ) and tau pathology and conversely, Aβ and tau aggregation disrupt sleep. The sleep/wake cycle is a master regulator of metabolic and neuronal activity, where daily oscillations in activity are coupled to the production and clearance of Aβ and tau. Although modulating neuronal activity alters both sleep/wake cycles and Aβ/tau release, less is known about how fluctuations in glucose metabolism drive changes in sleep and Alzheimer’s disease related pathology.
The goal of our work is 1) to determine whether changes in metabolic activity lead to changes in neuronal activity to disrupt sleep in Alzheimer’s disease and 2) whether metabolic dysfunction can serve as a novel therapeutic target to rescue sleep and Alzheimer’s pathology.
Using hippocampal biosensors coupled with EEG/EMG recordings, our work will investigate how glycemic variability and peripheral glucose intolerance affect sleep and Alzheimer’s disease in rodent models of Alzheimer’s related pathology. Moreover, we will establish whether normalizing peripheral glucose homeostasis through treatment with the diabetic medication, metformin, is sufficient to preserve and restore sleep architecture in the setting of Alzheimer’s disease.

KATP channel activity in Alzheimer's disease:
from pathology to treatment


Elevated blood glucose levels, or hyperglycemia, can increase brain excitability and amyloid-beta (Aβ) release offering a mechanistic link between type-2-diabetes and Alzheimer’s disease (AD). Since the cellular mechanisms governing this relationship are poorly understood, we explored whether ATP-sensitive potassium (KATP) channels, which couple changes in energy availability with cellular excitability, play a role in AD pathogenesis. First, we demonstrate that KATP channel subunits, Kir6.2/KCNJ11 and SUR1/ABCC8 are expressed on excitatory and inhibitory neurons in the human brain and cortical expression of KCNJ11 and ABCC8 changes with Alzheimer’s pathology in humans and mice. Next, we explored whether eliminating neuronal KATP channel activity uncoupled the relationship between metabolism, excitability, and Aβ pathology in a novel mouse model of cerebral amyloidosis and neuronal KATP channel ablation (e.g. APP/PS1, Kir6.2-/- mouse). Using both acute and chronic paradigms, we demonstrate that Kir6.2-KATP channels are metabolic sensors that regulate hyperglycemic-dependent increases in interstitial fluid levels of Aβ, amyloidogenic processing of APP, and amyloid plaque formation, which may be dependent on lactate release. These studies identify a new role for Kir6.2-KATP channels in Alzheimer’s disease and suggest that pharmacological manipulation of Kir6.2-KATP channels holds therapeutic promise in reducing Aβ pathology in diabetic or prediabetic patients.

Inward rectifying, ATP-sensitive potassium (KATP) channels are not restricted to neurons but found through the body. They also regulate excitability in 1) the vasculature, where they modulate vasodilation and vasoconstriction; and 2) pancreatic beta cells, where rising blood glucose triggers KATP channel closure and insulin release. Thus, KATP channel activity in different cell types cause different physiological effects.
Sulfonylureas are KATP channel antagonists and widely used anti-hyperglycemic medications to treat T2D. However, their effect on the neurovasculature and Alzheimer’s related pathology is unknown. Thus, we investigated the effects of the KATP channel antagonist, glyburide, on Alzheimer’s related pathology in a mouse model of Aβ overexpression (e.g. APPswe/PSEN1dE9 mice). We found that peripheral glyburide treatment: 1) decreased Aβ pathology; 2) reduced the activity dependent release of Aβ; and 3) altered the neurovascular response, reduced arterial stiffness, and normalized pericyte-endothelial cell morphology. These preliminary data suggest that KATP channels play a critical and previously unappreciated role linking Aβ with neurovascular dysfunction in AD. Ongoing work in the lab is exploring whether KATP channels are a druggable target for modulating Aβ and tau pathology in AD.
A novel approach for isolating extracellular vesicles from the hippocampal interstitial fluid (EV-ISF)

Brain-derived extracellular vesicles (EVs) play an active role in Alzheimer’s disease (AD), relaying important physiological information about their host tissues. The internal cargo of EVs is protected from degradation, making EVs attractive AD biomarkers. However, it is unclear how circulating EVs relate to EVs isolated from disease-vulnerable brain regions. We developed a novel method for collecting EVs from the hippocampal interstitial fluid (ISF) of live mice. EVs (EV-ISF) were isolated via ultracentrifugation and characterized by nanoparticle tracking analysis, immunogold labeling, and flow cytometry. Mass spectrometry and proteomic analyses were performed on EV-ISF cargo. EV-ISF were 40-150 nm in size and expressed CD63, CD9, and CD81. Using a model of cerebral amyloidosis (e.g. APPswe,PSEN1dE9 mice), we found protein concentration increased but protein diversity decreased with Aβ deposition. Genotype, age, and Aβ deposition modulated proteostasis- and immunometabolic-related pathways. Changes in the microglial EVISF proteome were sexually dimorphic and associated with a differential response of plaque associated microglia. We found that female APP/PS1 mice have more amyloid plaques, less plaque associated microglia, and a less robust- and diverse- EV-ISF microglial proteome. Thus, in vivo microdialysis is a novel technique for collecting EVISF and offers a unique opportunity to explore the role of EVs in AD.
Current Funding
-
NIA R01 AG068330 (PI)
-
NIA R01 AG061805 (CoI)
-
NIA R01 AG065839 (CoI)
-
BrightFocus Foundation (PI)
-
WF-TARC Pilot Grant (PI)
-
ADRC Pilot Grant (PI)
-
CTSI Pilot Grant (CoI)
-
NIA K01 AG050719 (PI)
-
ADRC Pilot Grant (CoI)
-
Donors Cure New Vision Award (PI)
-
NINDS F32 NS080320 (PI)
-
McDonnell Center for Systems Neuroscience Small Grant (PI: Macauley/Bauer)
-
Batten Disease Research and Support Association Research Fellowship
-
NINDS F31 NS056718 (PI)
Past Funding


Exosomes are small extracellular vesicles (EVs) that originate from the endosomal system and are secreted extracellularly. Exosome cargo is unique to their cell of origin and protected from degradation as the vesicles move throughout the circulation. Thus, exosomes act as messengers of pathological conditions and are being pursued as biomarkers in Alzheimer’s disease. To date, studies investigating exosome changes in AD have focused on exosomes isolated from the CSF, blood, or post-mortem brain tissue. However, these studies fail to fully capture 1) whether extracellular vesicle (EV) and exosome release into the brain’s ISF is impaired in AD in vivo, 2) how the progression of Alzheimer’s disease affects the exosome proteome in a brain region- and cell type-specific manner in vivo, 3) whether the ISF exosome population mirrors the blood or CSF pool, and 5) how exosome secretion from specific cell types, like neurons and glia, is altered in AD. Therefore, it is vital to uncover whether these changes in ISF-derived EVs are truly exosome specific, how the ISF population of exosomes changes during AD pathogenesis, and whether the local changes in ISF exosomes can be captured in the blood for biomarker development.
We leveraged our expertise in lysosomal biology with our technical expertise with in vivo microdialysis to develop a novel method for isolating exosomes from the hippocampal interstitial fluid (ISF) of unanesthetized, freely moving mice. Coupled with proteomic profiling, we can characterize brain region- and cell type-specific changes in exosomes relative to Alzheimer’s-like pathology in vivo. Moreover, we can identify unique proteins on the exosome surface that change with amyloid-beta (Aβ) or tau and explore their use as novel blood based biomarkers in AD. We feel this novel approach will yield novel insights into the role of exosomes in AD pathogenesis.